The post Perthes Disease appeared first on Prime Health Channel.
]]>Perthes disease is a rare kind of disability, primarily affecting the hip joint of children. It is characterized by the softening and ultimate breaking down of the top part of the thigh bone, called the femoral head. This form of osteochondritis is also called Calvé-Legg-Perthes disease after each of the three doctors who first described it in 1910.
What Causes Legg‑Calve‑Perthes disease
The condition occurs when small blood vessels suddenly stop supplying blood to the femoral head. What exactly causes this disruption is not known, but a number of studies show that certain factors may be associated with it such as:
- Blood Clotting: Disturbances in the formation of blood clot in the artery called, ligamentum teres femoris, may result in an insufficient blood supply to the femur.
- Direct Trauma: Blood vessel damage caused by trauma may affect the femoral head and hip joint.
- Constriction of Femoral Artery: This may lower blood supply to the medial femoral circumflex artery, the main blood vessel supplying blood to the femoral head.
Risk Factors
Age: Although the disorder may affect children of nearly any age, it is most common in between ages 4 and 8.
Gender: It is up to five times more common in boys than in girls.
Race: It is more common among Caucasian children.
Family History: In a few cases it seems to be hereditary, but there is a lack of evidence to prove it.
Socioeconomic Class: Its occurrence is high among those belonging to lower social classes.
Apart from these, certain diseases like transient synovitis, and some endocrine disorders may also lead to the condition.
Stages of Perthes disease
The lack of blood supply to the femur, results in a number of changes in the bone structure that show up in four clear stages.
- Necrosis: During this early stage, lasting for several months, the bone cells of the affected area die.
- Fragmentation: Occurring over a period of 1 to 2 years, the body replaces its dead bone with a softer one. This bone, being weaker, may fracture or become distorted easily.
- Re-ossification: In this longest stage, lasting for a few years, stronger new bone cells begin to grow in the femoral head.
- Healed: The bone growth is complete, with the femoral head attaining its final shape.
Complications
- Lower back pain
- ADHD
- Hinge abduction
Perthes disease Complication in Adults
- Hip arthritis
- Osteoarthritis
Signs and Symptoms
The earliest symptom constitutes a change in the manner a child walks or runs. The standard gait variations observed are:
- Occasional limping
- Stiffness and limited range of motion in the hip joint
- A peculiar running style
Another prominent symptom is pain, and it entails the following characteristics:
- Areas Affected: Either one hip (unilateral) or both hips (bilateral), groin, thigh or knee (known as referred pain)
- Features: Worsens with activity and relieves with rest
Other Symptoms
- Muscle spasms
- Shortening of the affected leg
- Thinner thigh muscles
- Fever
Depending upon a child’s activity level, the symptoms occur intermittently over a period of weeks to months, and mostly affect one hip. Both hips are affected in only 10% to 15% of all cases.
Diagnosis and Tests
The diagnosis generally involves:
- Physical examinations to assess a child’s range of movement in the hip
- X-rays of the hip joint
- Bone scan to determine the disease (if in the earlier phase, an X-ray picture comes normal, but symptoms persist)
- MRI scans to check the extent of damage.
- Blood tests to rule out problems like an infection and also to determine levels of calcium, vitamin D, and boron as these play a vital role in strengthening bones.
Differential Diagnosis
- Multiple epiphyseal dysplasia
- Spondyloepiphyseal dysplasia
- Sickle cell disease
- Gaucher disease
- Hypothyroidism
- Meyers dysplasia
Treatment and Management
Treatment mainly aims at restoring normal hip movement, ensuring that the femoral head remains well seated in the hip socket and relieving inflammatory symptoms. It can be treated by various means, but before treatment, these factors are evaluated:
The Age of the Child: As young children, (age 6 and below) tend to recover fast by growing new, healthy bones, thereby requiring no treatment.
The Extent of Damage to the Femoral Head: As the possibility of regrowth without deformity is reduced if the disorder damages more than 50% of the femoral head.
Older children and those with over 50% of damage usually need surgical intervention.
Nonsurgical Treatment for Small Children (2 to 6 years)
Observation and Physiotherapy
Firstly, a child is monitored with the help of x-rays so as to determine if the femoral head is regrowing properly. Then, he is given a home exercise routine to follow, with exercises like hip abduction and hip rotation that, focussing on internal rotation and reducing stiffness, may help in recovering the range of motion of the hip joint.
Anti-inflammatory Medications
Anti-inflammatory medicines, such as ibuprofen, and acetaminophen (Tylenol) may lessen inflammation. Depending on the stage of the disorder, these may be recommended for several months.
Crutches and Traction
Some children may require crutches for a short time to prevent too much pressure on the affected hip, while those with severe pain may need a period of bed rest. Traction is also done by placing pulling forces on the femoral head so as to relieve the tension existing between it and the hip bone.
Casting and Bracing
A leg cast is used in case a deformity is indicated by x-rays, or if the range of motion becomes restricted. At first, both legs need to be kept spread apart for nearly four to six weeks so that the femoral head remains in its place, within the hip socket (acetabulum). After this, hip flexibility is maintained by using a night-time brace. Physical exercises are resumed after the cast is removed. Leg braces ensure maximum mobility by providing external support to the hip and leg.
Orthoses
These orthopedic devices help to maintain a normal anatomic position of the leg and hips by promoting the femur’s internal rotation.
Follow up Measures to Take at Home
- Remodeling of Activity: High-impact actions, like running, bouncing or jumping should be avoided
- Administration of Heat or Cold: Hot packs or ice may help in relieving hip pain associated with the disease
Surgical Treatment for Older Children (above 8 years)
The three intended outcomes of surgery involve:
- Reestablishment of proper alignment of hip bones
- Restoration and maintenance of the femoral head
- Improvement of the shape of hip joint so as to prevent arthritis in later age
Osteotomy
This is carried out by cutting and repositioning the femoral or pelvic bone by changing the femoral head’s position or deepening the socket, using pins or plates, so that the femoral head remains within the acetabulum. After the procedure is over, a cast is used for 6 to 8 weeks to protect the joint.
Removal of Loose Bodies or Excess Bone
Torn flaps of cartilage or lose parts of bones around the femoral head are removed so as to ease movement as well as relieve pain.
Hip Distraction
In children with severe symptoms, a device, known as external fixator is attached to the outside of the hip through a process called distraction osteogenesis. This device reduces joint compression, thereby allowing it to heal without damaging tendons or ligaments. Since stability is provided after being kept in position for three to six months, children can walk, even wearing the external fixator.
Hip Replacement Surgery
In this surgery, the hip ball is replaced within the socket.
According to several studies, the disability may also be controlled through Ayurvedic treatment, but more research needs to be done to validate this.
Prognosis and Long Term Effects
With regrowth of the femoral head, the hip joint becomes normal in most cases, starting to work well within about two years. However, if it does not reform properly and attains a flat shape, stiffness or other hip problems may happen later in life.
Can Perthes disease be Prevented
The disability cannot be prevented, but certain measures can be taken to maintain a proper bone health:
- Optimum Exposure to Sunlight: Exposing one’s hands and face to sunlight for about 15 minutes per day, naturally influences the body to make vitamin D; the chief nutrient that ensures calcium is absorbed and employed by the body.
- Healthy Nutrition: Having a healthy diet is necessary for maintaining proper weight and minimising bone loss.
Incidence
It is estimated that this disorder affects 1 in 10,000 children per year.
Perthes disease ICD-9-CM Codes and ICD-10-CM Codes
The ICD-9-CM code of Perthes disease is 732.1, and the ICD-10-CM code is M91.1.
The post Perthes Disease appeared first on Prime Health Channel.
]]>The post Jacobsen Syndrome appeared first on Prime Health Channel.
]]>Jacobsen syndrome or JBS, named after the Danish physician Petra Jacobsen who first identified and described it 1973, is a rare condition characterized by the loss of genetic material from the tip of the long arm (q) of chromosome 11. Hence, the condition is often called chromosome 11q deletion syndrome/disorder or partial 11q monosomy syndrome. The deletion size usually varies between 7 and 20 Mb while larger deletions may lead to disability to think and remember, behavioral issues, and other congenital defects.
What Causes Jacobsen Syndrome
It occurs due to the loss of several genes at the terminus of chromosome 11, which in most cases is brought about by a spontaneous mutation in parental sperm or egg, a random error in the formation of gametes, or a fault in cell division during fetal development. This deletion or loss of genetic materials is termed as de novo deletion. But, the triggering factors for such genetic changes are yet to be known.
Can it be Inherited
In rare cases, the deletion is inherited from a parent having balanced chromosomal translocation, a condition in which the genetic material is retained within the chromosome 11 even after the breakage of a part of the chromosome, which prevents any detectable abnormalities. In such cases, the parent is not affected by the disorder. However, a person with Jacobsen syndrome can also pass it on to his/her descendants.
Risk Factors
- Gender: Females are more at risk to develop the condition than males. The ratio of its occurrence between the two sexes is 2:1.
- Family history: About 15% of the people inherit Jacobsen syndrome from their parents.
Can Jacobsen Syndrome be Prevented
No specific guidelines exist for the prevention of Jacobsen syndrome. However, periodic medical screening tests may be performed if there are past occurrences in family members. Prenatal genetic testing of both the expectant parents and the fetus may be done to assess the risks during pregnancy.
Signs and Symptoms
Individuals with JBS may show wide-ranging symptoms, which are more severe in the case of larger deletions than the smaller ones. The following signs and symptoms have been reported in patients:
- Platelet dysfunction, pancytopenia or thrombocytopenia
- Speech and motor developmental delay
- Cognitive impairment along with learning difficulties
- Distinct facial features such as a large head (macrocephaly), prominent forehead (trigonocephaly), droopy eyelids (ptosis), wide-set eyes (hypertelorism), small, low-set ears, broad nasal bridge, upper eyelid skin fold (epicanthal folds) at the inner corner of the eyes, small lower jaw, and a thin upper lip
- Short height
- Sinus, ear infections, and hearing loss
- Feeding difficulties during early childhood
- Skeletal abnormalities
Complications
- Combined immunodeficiency characterized by recurrent infections
- Paris-Trousseau syndrome, a disorder of blood platelets, causing easy bruising and abnormal bleeding
- Attention-deficit hyperactivity disorder with behavioral issues like poor concentration and easy distractibility
- Autism characterized by inappropriate social interaction and impaired communication skills
- Compulsive behavior like shredding paper
- Congenital heart defects
- Kidney problems like double ureters, single kidney, and cysts in children
- Gastrointestinal disorders including pyloric stenosis, intestinal obstruction, constipation, and malformations of the GI tract affecting children
How is Jacobsen Syndrome Diagnosed?
After a complete physical examination, the family history of the patient is evaluated, and then the symptoms are assessed. Clinical tests to find out any intellectual deficit, or facial abnormalities, and low blood platelet count are used for making a diagnosis.
MRI, CT, or cerebral ultrasound may be done for finding out abnormalities of the brain. Laboratory findings are confirmed by cytogenetic analysis, including CGH+SNP microarray analysis.
Diagnosis in expecting parents involves procedures for analyzing samples of amniotic fluid or chorionic villus.
Differential Diagnosis
- Thrombocytopenia (platelet abnormality) caused by sepsis
- Turner syndrome
- Noonan syndrome
- Kabuki syndrome with symptoms like short stature, mental retardation, and abnormal palpebral fissures
Treatments for Jacobsen Syndrome
While there is no known cure for the condition, treatment is based on the symptoms present, with options including the following:
- For people affected by low platelet count, regular monitoring along with blood transfusions before or during surgical procedures may be required.
- Eye abnormalities and other visual problems may be improved with contact lenses, glasses, and surgery.
- Malformations of the kidney, urinary tract, and gastrointestinal tract, as well as abnormalities of bones, joints, muscles, and tendons, may be treated with surgery.
- Individuals affected by congenital heart defects may be treated with different medications. Infections of the respiratory tract or the heart’s valves and lining require high doses of intravenous antibiotic treatment before surgery.
- Patients with coordination and mobility issues may be treated with physical therapy for improving balance, posture, and strength.
Prognosis and Quality of Life
Medical therapy can address health complications and improve the overall health of an affected child, thus helping him or her to reach proper developmental milestones. Life expectancy of children born with Jacobsen syndrome is unknown. Some may die in their neonatal stage due to excessive bleeding and severe heart malformations while others may live into adulthood. The person recorded as having lived the longest with JBS lived for over 45 years of age. Finding support groups can help the parents and their child cope with the life threatening condition.
Incidence and Prevalence
It is estimated that 1 out of 100,000 newborns have chances of developing Jacobsen syndrome. According to recent population-based studies, over 200 individuals have been affected by this genetic condition.
ICD-9-CM and ICD-10-CM Codes
The ICD-9-CM code for JS is 758.3 while its ICD-10 code is Q93.5.
The post Jacobsen Syndrome appeared first on Prime Health Channel.
]]>The post Cri du chat appeared first on Prime Health Channel.
]]>Cri du chat is a rare genetic condition [1] that occurs when a part of chromosome number 5 is absent.
It is also referred to as:
- Chromosome 5p deletion syndrome
- 5p minus syndrome
- Lejeune’s syndrome
Cri du chat History
The syndrome was first described in 1963 by Jerome Lejeune, a French geneticist and pediatrician.
Cri du chat Epidemiology
It is estimated to affect one in every 20,000-50,000 newborns [3]. The disease is reported in people of all ethnic backgrounds. The frequency is greater in girls. The female to male ratio of this disorder is 4:3.
Cri du chat Symptoms
Its signs and symptoms include:
- High-pitched or weak crying sounds, similar to that of a cat
- Small head (microcephaly)
- Low birth weight
- Rounded face
- Small jaw (micrognathia)
- Eyes placed wide apart
- Broad, flattened nose bridge
- Skin folds above the eyelids
- Malformed ears
- Palatal abnormalities, characterized by high or unusually narrow palates
- Intellectual disability
- Feeding problems, due to difficulties in sucking and swallowing
- Behavioral problems, like aggression, hyperactivity, repetitive movements and tantrums
- Weak muscle tone (hypotonia) in infancy
- Retarded or incomplete motor skill development
- Single line in the palm
- Downward slant to the eyes
- Renal malformations, such as horseshoe kidneys (rare)
Cri du chat Causes
It arises due to deletion of the short arm-end of chromosome number 5. This change is referred to as 5p-. The size of the deletion differs from one patient to another. As per studies, developmental delay and intellectual disability are more acute in larger deletions than in smaller ones. According to researchers, the loss of a particular gene CTNND2 is linked with acute intellectual disability.
The majority of cases are supposed to arise during development of the sperm or the egg. A small number of cases arise due to passing of a varying, rearranged form of the 5th chromosome from parents to baby.
It is still unclear as to how the loss of genes results in the problems associated with Cri du chat.
Cri Du Chat and Inheritance
Most cases are not inherited. Only around 10% patients inherit the chromosomal abnormality from one unaffected parent. In such cases, the parent is a carrier of a balanced translocation (a chromosomal rearrangement) in which no genetic component is lost or gained.
Typically, victims do not have any family history of the syndrome. The deletion usually occurs randomly during early stages of fetal development or during the formation of eggs or sperm.
Cri du chat Diagnosis and Testing
The condition is generally diagnosed at birth. Doctors discuss with parents about the characteristics in their infants. Diagnosis usually begins with a physical exam, which may reveal:
- Poor muscle tone
- Inguinal hernia
- Diastasis recti (separation of belly muscles)
- Problems with the folding of the outer ears
- Epicanthal folds, additional skin folds above the inner corner of the eye
X-rays of skull may show possible problems with the basal shape of skull. Genetic tests can reveal deletion of a section of chromosome 5 [2]. The deletion may also be revealed through amniocentesis [4] or analysis of chrionic villi samples of carriers.
Ultrasound scans may also be required in some cases.
Cri du chat Treatment
There is no particular cure. Physicians only suggest ways for symptomatic treatment or management. Parents of sufferers can visit genetic counselors to conduct testing and determine which one of them is having a change in chromosome 5. Adults with a family background of Cri du chat should undergo genetic testing.
Heart defects [5] often need surgical correction. After leaving hospital, regular visits to health care providers are necessary.
Cri du chat Prognosis
The outcome is mixed, as there is no specific cure. Half of all sufferers learn enough verbal skills to interact as normally as possible, despite impaired language development. The cat-like cry gets less apparent with time. With physical therapy and consistent educational intervention, quality of life is highly improved [6]. However, intellectual disability is commonly noticed.
Cri du chat Complications
The complications are based on the amount of symptoms and intellectual impairment. Physical and mental symptoms may affect the level of dependence of sufferers on themselves.
Can Cri du chat Kill You?
Majority of sufferers have a normal life expectancy. Life span can be reduced in a small number of patients with severe organ defects or other life-threatening problems. It is not fatal in most cases.
Cri du chat Prevention
No specific way is known to prevent this syndrome. Couples with a family history of Cri du chat, and planning pregnancy, may consider genetic counseling.
Cri Du Chat Support Groups
Although it is a rare disorder, there are various support groups that offer guidance and information to parents of Cri Du Chat sufferers. Parents may contact such organizations as:
Genetic Science Learning Center
University of Utah
383 Colorow Dr.
Salt Lake City, Utah 84108
Phone: (801) 585-3470
Cri du Chat Syndrome Support Group [7]
5 Latimer Drive
Steeple View
Laindon
Essex
SS15 4AD
Tel: 0845 094 2725
Web: www.criduchat.org.uk
Cri du Chat Support Group of Australia Inc.
Association number A0033602W
104 Yarralumla Drive, Langwarrin, VIC, 3910, Australia
Phone or Fax: 61 3 9775 9962 (Wendy)
Cri du chat Pictures
The following images show the abnormal facial features of Cri du chat patients.
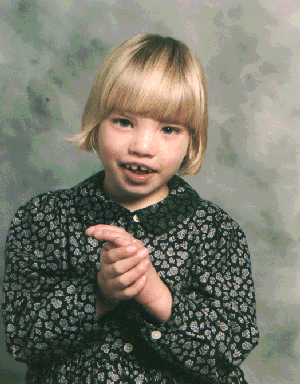
Picture 1 – Cri du chat
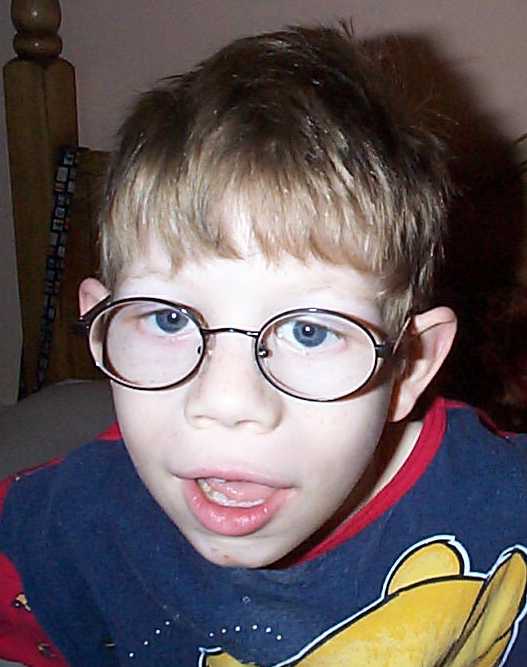
Picture 2 – Cri du chat Image
Cri du chat ICD9 Code
Its ICD9 Code is 758.31.
The post Cri du chat appeared first on Prime Health Channel.
]]>The post Craniodiaphyseal dysplasia appeared first on Prime Health Channel.
]]>Craniodiaphyseal dysplasia is a very rare genetic disorder of the bone [1]. Only around 30 cases of its have been reported so far. It is also known as lionitis or simply as CDD.
Craniodiaphyseal dysplasia History
Cases with features similar to CDD were first reported in 1949 (by Halliday) and in 1958 (by Joseph et al). It was then regarded as a type of Camurati-Engelmann disease. It was not until in 1969 that it was first described by Gorlin et al as a very acute bone disease characterized by massive hyperostosis and sclerosis, particularly affecting the facial and skull bones.
CDD became known through the 1985 drama film “Mask” by Peter Bogdanovich that showed the case of an American boy Roy L. Dennis aka Rocky who died of the disease in 1978.
Craniodiaphyseal dysplasia Symptoms
There is buildup of calcium within the skull, which reduce the size of cranial foramina. This leads to abnormal facial features and lowers the lifespan of patients. The calcium deposits can also reduce the hole in the cervical spinal canal. There is compression of cranial nerves, the intracranial contents and the foramen magnum, which commonly results in:
- Blindness
- Epilepsy
- Facial diplegia
- Loss of hearing
- Mental retardation
Stenosis of the cervical canal may cause quadriparesis (a late complication of hyperostosis). Sclerosis of the expanded diaphyses and hyperostosis can be observed in the long bones. In some cases, a postural defect in the metaphyses may also be noted.
Craniodiaphyseal dysplasia Causes
It is supposed to be hereditary, although the underlying genetics is unclear. The disease is acquired in an autosomal recessive fashion. Very few cases of CDD have been described, which causes difficulty in determining genetic association.
Craniodiaphyseal dysplasia Diagnosis
A difference in the size of eyes is a common reported sign for CDD. Physicians also look for clinical symptoms like:
- Dacryocystitis [2]
- Seizures
- Mental retardation
- Paralysis
These are complications ensuing as a result of diminutive foramina. X-ray images can show regions of abnormal bone density, where excess calcium deposition occurs and many osteoblasts and cells that manufacture new bones are present.
Craniodiaphyseal dysplasia Differential Diagnosis
Mutation analysis of the gene TGFB1 lets doctors rule out the presence of Camurati-Engelmann disease, the signs of which are similar to those of CDD. Other disorders to be considered include:
- Craniotubular dysplasias
- Van Buchem’s Dysplasia
Craniodiaphyseal dysplasia Treatment
CDD sufferers, particularly those with tracheal intubation problems and difficulties with airway management, need to be taken to an anesthetist. Decompressing craniectomy can be used to enlarge the middle and the anterior fossae in case of acute brain compression with signs of elevated intracranial pressure.
In case of papilledema, decompression of optic and orbital nerve may be needed for cure. Calcitriol/calcitonin therapy, along with prednisone or low calcium diet may change the clinical course of progression [3].
Long-term treatment is based on controlling rapid malformation of bones.
Craniodiaphyseal dysplasia Management and Control
Further progression of CDD can be managed by close observation of the health of sufferers. Magnetic Resonance Imaging (MRI) should be used as a routine component for evaluating patients.
Craniodiaphyseal dysplasia Prognosis
The outcome is poor. Generally, benefits from dacryocystorhinostomy, craniofacial remodeling and choanal stenosis surgery persist only for a short time. Reconstitution of osseous optic canals occurs rather rapidly, and there may be a recurrence of papilledema. Most patients have been found to die in childhood in the few reported cases of CDD. Life expectancy is extremely bad for patients with this disease. It is vital to recognize these conditions on an early basis as therapy directed at the underlying bony defect can succeed the best if started during infancy.
Craniodiaphyseal dysplasia Complications
The probable complications include:
- Affected vascular supply (due to hyperostosis)
- Severe facial deformity
- Elevated numbers of osteoblasts
- Loss of vision [4]
- Deformation of ossicles due to bony overgrowth
- Significant brain compression
- Epilepsy
- Complete hearing inability
- Reduced air spaces of mastoid, middle ear cavity and external auditory canal by hyperostotic bone
- Facial diplegia
- Dysfunction of facial nerves
- Problems in air conduction, due to anomalous ossicles with hyperostosis
- Moderate sclerosis and postural defects (in clavicles, ribs and pelvis)
- Retarded mental abilities
- Quadriparesis
- Mechanical damage of nerve fibers
- Nasolacrimal obstruction
Craniodiaphyseal Dysplasia Support Group
For information and guidance, family members of CDD sufferers may contact:
Office of Rare Diseases Research
National Center for Advancing Translational Sciences (NCATS)
National Institutes of Health
6701 Democracy Boulevard
Suite 1001, MSC 4874
Bethesda, MD 20892
For courier use (Bethesda, MD 20817)
(301) 402-4336 (voice)
(301) 480-9655 (fax)
Email: [email protected]
Craniodiaphyseal dysplasia Pictures
Check these photos and know about the physical appearance of patients with CDD from the images.
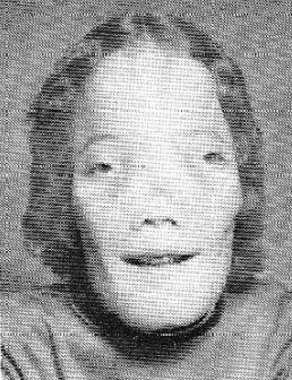
Picture 1 – Craniodiaphyseal dysplasia

Picture 2 – Craniodiaphyseal dysplasia Image
Craniodiaphyseal dysplasia ICD10 Code
The ICD10 Code for this disorder is M85.2.
References:
Craniodiaphyseal dysplasia (Wikipedia)
Craniodiaphyseal dysplasia (Piczo.com)
Craniodiaphyseal dysplasia (Orpha.net)
What is Craniodiaphyseal dysplasia? (Wisegeek)
The post Craniodiaphyseal dysplasia appeared first on Prime Health Channel.
]]>The post Cornelia de lange syndrome appeared first on Prime Health Channel.
]]>Cornelia de lange syndrome (CdLS) is a congenital defect [1] that is not always detected at birth.
It is also known by the names:
- Amsterdam dwarfism
- Bushy Syndrome
Cornelia de lange syndrome – Epidemiology and Incidence
It is estimated to affect 1 out of every 10,000 live births. The exact incidence is unknown, although it is assessed to be 1 in 10,000-30,000 live births.
Cornelia de lange syndrome Symptoms
It is characterized by:
- Distinctive facial appearance [2]
- Difficulties in feeding
- Behavior problems (compulsive behaviors, daily aggression, hyperactivity, sleep disturbance, self-injury, social anxiety)
- Genitourinary problems (cryptorchidism, hydronephrosis, hypogonadism, hypoplasia, renal dysplasia, subcortical renal cysts, impaired renal function, urethral reflux)
- Growth deficiency (prenatal and postnatal), or retarded growth before and after birth
- Psychomotor delay, generally manifested by acute to profound intellectual disability
- Developmental defects (primarily involving the upper extremities – arms and hands)
- Autism (in some patients), marked by impaired social interaction and communication
The facial abnormalities involve:
- Arched eyebrows, often growing together in the centre (synophrys)
- Small, upturned nose and thin lips (downturned)
- Low-set ears
- Long eyelashes
- Dental problems (small, widely spaced teeth)
Additional symptoms include:
- Abnormally small head (microcephaly)
- Cleft palate
- Low birth weight and height (generally less than 2.5 kg/5 pounds)
- Digestive tract problems (Pyloric stenosis, Gastro-esophageal reflux, alimentary canal abnormalities)
- Excessive body hair or Hypertrichosis (hypoplastic nipples and umbilicus, hirsutism on the back, long eyelashes)
- Eye problems (ptosis, myopia and nystagmus)
- Central nervous system issues
- Congenital heart defects (atrial septal defect, Fallot’s tetralogy pulmonary stenosis and ventricular septal defect)
- Hearing problems (Hearing loss, stenosis of the external auditory canals)
- Heat intolerance
- Lack of pain sensation
- Short height
- Seizures
- Skeletal abnormalities (hypoplastic or absent ulna, Oligodactyly, clinodactyly, single palmar crease, syndactyly, proximal placement of thumb/thumbs)
- Respiratory problems (upper respiratory tract infections, pneumonia, bronchopulmonary dysplasia)
Cornelia de lange syndrome Causes
Most cases occur from spontaneous mutations in the genes SMC1A, SMC3 and NIPBL. NIPBL mutations have been spotted in over half of all CdLS sufferers. Cases involving mutations of the two other genes are much less common. [3]
Mutations in the three genes can disrupt gene regulation during critical early developmental phases. As per studies, mutations in the SMC1A and SMC3 genes cause mild cases (milder symptoms) than those in cases of NIPBL mutations. Most cases involving SMC1A mutations arise in people without any family history of the disease.
SMC3 or NIPBL mutations are supposed to be inherited in an autosomal dominant pattern. Only one copy of the mutated gene can cause CdLS. An X-linked inheritance pattern is associated with SMC1A mutations. In other words, the responsible mutated gene is located on X chromosome (one of the two sex chromosomes). X-linked cases of CdLS affect both males and females similarly.
Cornelia de lange syndrome Diagnosis
Diagnosis is based on:
- Symptoms manifested by sufferers
- Medical history
- Physical examination
- Laboratory testing
Recommendation to a genetic specialist can also help in confirmation.
For parents of CdLS sufferers wanting to have more kids, prenatal ultrasound can help assess subsequent pregnancies. In some organizations, ultrasound is deemed as best for prenatal diagnosis.
Cornelia de lange syndrome Differential Diagnosis
It includes isolating the signs of CdLS from those of fetal alcohol syndrome and other rare genetic conditions.
Cornelia de lange syndrome Treatment
Cure involves:
Non-surgical approach
Early medical intervention helps improve:
- Feeding problems
- Urinary system anomalies
- Congenital heart disease
- Visual and hearing impairment
Medical intervention can include:
- Emphasis of perceptual organizational tasks
- Computer programs, to stress visual memory
- Stressing fine motor activities in education
- Tactile stimulation, during indirection
Surgical approach
In some cases, surgery (under anesthesia effects) may be required for correction of:
- Cleft palate
- Gastroesophageal reflux disease
- Hip dislocations
- Intestinal malrotation/volvulus
- Lacrimal duct stenosis
- Nasal polyps
- Pyloric stenosis
- Undescended testis
Consultations
Guardians can consult the following specialists:
- Cardiologists
- Gastroenterologists and nutritionists
- Geneticists
- Hearing specialists
- Nephrologists (for congenital abnormalities, impaired renal functions or recurrent urinary tract infections)
- Neurologists
- Ophthalmologists
Cornelia de lange syndrome Management
Management is primarily based on specialists. General practitioners ensure overall health supervision and regular hospital reviews and recommend genetic counseling. Recurrence risk is reduced for spontaneous mutations.
Cornelia de lange syndrome Prognosis
Most premature deaths occur in acutely affected neonates, within the first 2 years of life [4]. A slightly reduced life span is observed in survivors. Life expectancy depends on the general health as well as the level of care.
Cornelia de lange syndrome Support Group
The Cornelia de lange syndrome (CdLS) Foundation, which is a non-profit Connecticut-based organization, provides support to family members. It helps in early and proper diagnosis of CdLS and encourages research into the causes and symptoms of the disease.
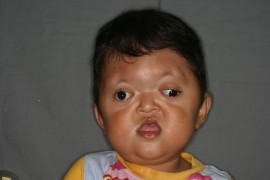
Cornelia de lange syndrome Pictures
The images show the appearance of CdLS sufferers.
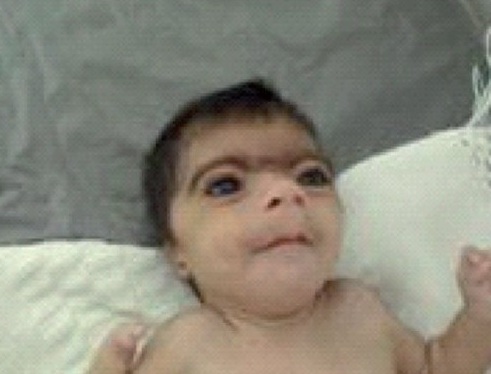
Picture 1 – Cornelia de lange syndrome
Picture 2 – Cornelia de lange syndrome Image
Cornelia de lange syndrome ICD9 Code
The ICD9 Code for CdLS is 759.89.
References:
- Cornelia de lange syndrome (Wikipedia)
- Cornelia de lange syndrome (Medscape)
- Cornelia de lange syndrome (nlm.nih.gov)
- Cornelia de lange syndrome (Patient.info)
The post Cornelia de lange syndrome appeared first on Prime Health Channel.
]]>The post Craniosynostosis appeared first on Prime Health Channel.
]]>This is a congenital defect in which one or more joints in between the bones in the skull of a newborn close even before the brain has fully developed [1]. This changes the pattern of growth of the infantile skull.
It is also known as “Premature closure of sutures”.
Craniosynostosis Epidemiology
It affects one out of 2,000 to 2,500 live births around the world.
Craniosynostosis Types
It is classified into the following forms:
- Sagittal synostosis (Scaphocephaly) – It affects the main suture at the apex of head (sagittal suture) and results in a broad forehead. The head grows long and narrow, rather than wide. The most common form of Craniosynostosis, it affects boys more than girls.
- Frontal plagiocephaly – The second-most common form, it affects girls more, and impacts the suture which runs from one ear to another.
- Coronal synostosis – It involves premature fusion of one of the coronal sutures, structures running from ears to the sagittal suture at the apex of the head. It may cause flattening of affected head section, slanted skull, deviated nose and elevated eye socket.
- Metopic synostosis – This is rarer, but may vary from mild to acute in intensity. It affects the metopic suture located near the forehead and leads to an abnormal head shape (Trigonocephaly).
Lamboid synostosis is a rare form that involves the lamboid suture, which runs across the skull, and is characterized by flattening of the affected head section.
Craniosynostosis Causes
It is not known what exactly causes this disease. However, it is believed to be hereditary in nature. No family history of the disorder is generally observed [2]. However, a hereditary form exists that occurs along with other problems like blindness, reduced intelligence and seizures.
Some genetic disorders commonly related with Craniosynostosis include:
- Apert
- Carpenter
- Crouzon
- Chotzen
- Pfeiffer syndromes
Most affected children have normal intelligence and are healthy otherwise.
Craniosynostosis Risk Factors
Individuals with a family history of the condition are at a high risk of suffering from it. This is true in case of the type of Craniosynostosis that affects members of the same family.
Craniosynostosis Symptoms
The signs and symptoms depend on the form of the disease that one suffers from, and typically include:
- Abnormal shape of head [3]
- Elevated hard ridge along the sutures that are impacted
- Reduced or non-enlargement of head size over time
- Absence of fontanelle (soft spot) on the skull of newborns
Some types of the disorder can be related with strabismus, a disorder in which the eyes fail to move in tandem.
Craniosynostosis Testing and Diagnosis
It initially involves a physical examination, in which the skull is felt. The following tests may be performed:
- X-ray exam of the skull
- CT scan of the head
- MRI scans (in isolated cases)
- Measurement of head width
- Ultrasound, that helps rule out Craniosynostosis in clinically suspected cases
Genetic Testing
It may help detect the disease if an underlying hereditary disorder is suspected as the cause. Although a blood sample is usually required, a sample of skin, hair or other tissue may be needed depending on the type of suspected abnormality. The sample may be sent for lab tests.
Craniosynostosis Treatment
While mild cases may require no surgery or medical intervention other than a cranial helmet (for reshaping the head), more severe cases need surgery as primary mode of cure. The timing and type of surgery depends on the type of the disease as well as the possible presence or absence of an underlying disorder.
Operative options include:
- Traditional surgery – An incision is made into the cranial bones and scalp to re-shape the affected skull section. Post-surgery, infants need at 3 days of hospital stay.
- Endoscopic surgery – It is a less invasive form of surgery, involving the insertion of a lighted tube (endoscope) through incisions made over the affected suture. It generally includes less bleeding, inflammation and hospital stay.
Craniosynostosis – Post-Operative Management
Following operation, the head of affected children are commonly wrapped with turban. Typically, they are shifted to an ICU setting for close monitoring after surgery. Possible complications like vomiting, headaches, fever and fatigue should be promptly evaluated.
Craniosynostosis Prognosis
The outcomes depend on the overall health of infants as well as the number of sutures (immovable joints) involved. Affected children operated on generally show good recovery. Quality of life especially improves when no genetic syndrome is associated.
The disease also has a risk of recurrence, which can range from 2-50%, and can only be determined after complete genetic assessment.
Craniosynostosis Complications
Patients may suffer from head deformity that can be acute and permanent if left untreated [4]. Other possible problems (which can involve long term effects) include:
- Seizures
- Developmental delays
- Learning disabilities and vision problems, in later years
- Raised intracranial pressure, causing irritability, vomiting , headaches, breathing difficulties etc.
Differing appearance may also result in psychological problems in adulthood. Possible risks involved with surgical repair include:
- Brain infection
- Brain inflammation
- Damage to brain tissue
- Fusion of bones again, requiring further operations
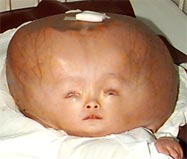
Craniosynostosis Pictures
The images here show how affected infants and adults look like.
Picture 1 – Craniosynostosis
Picture 2 – Craniosynostosis Image
Craniosynostosis ICD9 Code
The ICD9 Code for this disorder is 756.0.
References:
- Craniosynostosis (nlm.nih.gov)
- Craniosynostosis (Mayoclinic)
- Craniosynostosis (Wikipedia)
- Craniosynostosis (nhs.uk)
The post Craniosynostosis appeared first on Prime Health Channel.
]]>The post Calciphylaxis appeared first on Prime Health Channel.
]]>It is a syndrome that is a combination of vascular calcication, skin necrosis and thrombosis. A rare and serious disorder, it is characterized by systematic medial calcification of the arterioles which leads to ischemia and subcutaneous necrosis. This is a type of extra-osseous calcification which may occur in patients who have developed end-stage renal disease. In medical history, it has traditionally been classified as metastatic calcification which is an indication passive mineralization of serum calcium and phosphate crystals.
This syndrome usually arises in people who have undergone hemodialysis or have received a renal transplant.
Calciphylaxis Symptoms
This fatal disease is caused due to the deposition of calcium from blood vessels into the skin which leads to hypersensitivity. Some signs and symptoms related to Calciphylaxis are:
- Systemic medial calcification of the arteries.
- Formation of red papules, plaques and other nodules.
- Small vessel mural calcification with or without extravascular calcification, endovascular fibrosis and vascular thrombosis, all of which lead to tissue ischemia.
Other Symptoms
Some other problems related to this disease include:
- Painful skin ulcers
- Organ failures
- Bleeding from the scarred area
- Itching and burning sensations
- Purple or red colored lesions on abdomen, stomach or buttocks or thighs.
Calciphylaxis Causes
The cause of this syndrome is still not clearly known. It is not an allergic reaction but there are several additional factors that lead to its development.
Most cases of Calciphylaxis have been found to occur when there is a chronic renal failure, hyperparathyroidism or abnormal calcium-phosphate homeostasis. However, few case reports also suggest that the condition can occur even when there is no renal disease and a patient suffers from rheumatoid arthritis and Crohn disease.
Calciphylaxis mostly occurs in body areas having maximum fat. Fatty areas are at greater risk of developing thrombosis due to low blood flow or due to increased tendency for vascular kinking. Patients who are at risk include those who are obese and those who are exposed to immunosuppressive agents like glucocorticoids. People who are suffering from diabetes mellitus are also at risk of developing this rare disorder.
Know about some disorders associated with the development of Calciphylaxis:
- Systemic inflammation is a pre-disposing factor.
- Speculative associations include iron dextran infusion, aluminum toxicity and coagulation abnormalities.
- Several associations like hypercalcemia, vascular calcification, hyperphosphatemia and elevated calcium-phosphate product.
Calciphylaxis Diagnosis
The diagnosis for this disorder involves considering the medical history of sufferers as well as conducting a few physical exams, which include:
Blood Tests
Blood sample of the patient is taken to measure a variety of substances in the bloodstream, such as:
- Calcium
- Aluminum
- Urea
- Phosphorus
- Albumin
- Parathyroid hormone
- Creatinine
This test helps doctors to assess the functions of the kidney and liver. If creatinine, a chemical waste, does not leave your body through urine it indicates that the kidney is not functioning properly. Also, if there is excess of urea nitrogen found in blood, it is a precursor for this disease. Healthy liver function is detected by the presence of proteins like albumin and clearing the waste product of blood bilirubin. Presence of bilirubin in the blood means the liver is not functioning properly.
Deep Skin Biopsy
A tissue sample from the affected area of the skin is taken by a doctor to confirm whether the underlying condition is Calciphylaxis or any other infection.
Imaging Studies
Imaging studies like X-rays show a clear picture of the vascular calcifications that appear like branches is a common phenomenon in Calciphylaxis. Other additional imaging exams include mammograms, bone scans and high resolution CT scans.
Calciphylaxis Treatment
There is no specific treatment for Calciphylaxis. The optimal curative process for this disorder is prevention. However, following treatment approaches are accepted:
Intensive wound treatment
In this method of treatment, damaged tissues are removed surgically and also by other methods. In some situations, the damaged tissues are removed with the help of wet dressings, whirlpool treatments etc. Medications are also prescribed to reduce pain during wound care. Antibiotic treatment is part of treating and preventing wound infection.
Reduction of Calcium Deposits
Reduction of calcium deposits in the arteries can be carried out by the following methods:
Surgery
If a doctor finds out an overactive parathyroid gland as the reason behind over production of calcium, he may decide to remove the entire gland or only a part of it.
Dialysis
A doctor may also recommend continuing kidney dialysis and adjust their frequency along with the medications required with it.
Medications for Reducing Calcium Levels
The doctor will make an evaluation of the current medications and try to remove the potential factors that trigger Calciphylaxis. He may change the dose of calcium and vitamin D supplements and ask you to stop taking corticosteroids or iron. Doctor may also suggest a medication known as cinacalcet which helps control parathyroid hormone. Other medications, which are also recommended, include sodium thiosulfate (to flush out calcium through urine) and medications that improve the balance of calcium and phosphorus in the body.
Restoration of Oxygen and Blood flow
Doctors may suggest a hyperbaric oxygen therapy to maximize the amount of oxygen in the affected parts. Medications that thin out the blood may also be prescribed to prevent additional blood clots from forming. Low-dose tissue plasminogen activator or TPA may also be used by the doctor to help dissolve the blood clots inside the tiny blood vessels of the skin.
Calciphylaxis Pictures
Here are a few pictures to help you understand the disorder and its effects.
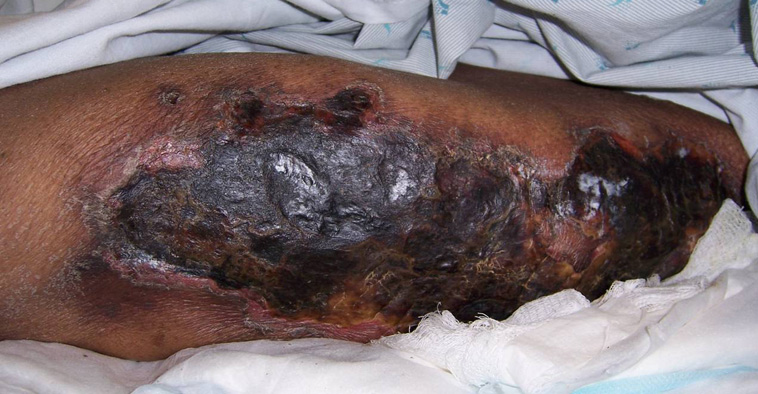
Picture 1 – Calciphylaxis
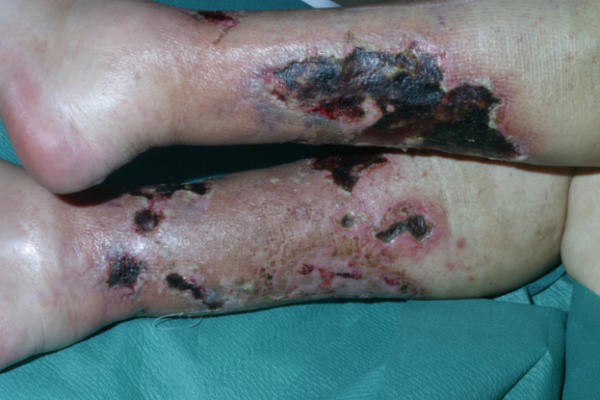
Picture 2 – Calciphylaxis Image
References:
http://www.uptodate.com/contents/calciphylaxis
http://en.wikipedia.org/wiki/Calciphylaxis
http://emedicine.medscape.com/article/1095481-clinical#a0218
http://www.health-issues.org/skin-disorders/calciphylaxis.htm
http://www.mayoclinic.org/calciphylaxis/treatment.html
The post Calciphylaxis appeared first on Prime Health Channel.
]]>The post Patau Syndrome appeared first on Prime Health Channel.
]]>Patau Syndrome Definition
It is a condition in which certain or all of the cells of the body of a person contain an additional genetic material from chromosome 13. In an individual with this disorder, the chromosome 13 appears thrice instead of twice.
The disorder is also referred to as Trisomy 13, although it is actually the name of one type of Patau Syndrome. Other names for this condition include:
- Trisomic
- Trisomee
- Trisomie 13
- Trisome
- Patau’s Syndrome
- Fetal Aneuploidy
- Trisomy 13 Syndrome
- Trisomy D Syndrome
Patau Syndrome ICD9 Code
The ICD9 Code for this disorder is 758.1.
Patau Syndrome Incidence
This is an extremely rare disorder. The condition has an incidence of one in every 8,000-12,000 live births. Significant geographic or racial differences in frequency are not evident. Less than 200 babies have been detected with the condition in the U.K.
The condition has been reported to have a greater prevalence in females, possibly due to low rate of survival among males. The disorder is completely evident at birth and is expressed prenatally. It may affect people from any ethnic background.
Patau Syndrome Symptoms
Those who suffer from this disease a number of times experience problems like:
- Heart defects
- Cleft lip or palate
- Reduced muscle tone
- Additional toes or fingers
- Small or poorly developed eyes
- Abnormalities of the brain or spinal cord
Babies with this condition do not gain weight or grow in stature as much as they should. They may have problems in being fed and also suffer from episodes where they have ‘apna’ or a temporary stoppage of spontaneous breathing.
Some other features of this disorder include:
- Seizures
- Small eyes
- Hernias
- Small head
- Scalp defects
- Clenched hands
- Low-set ears
- Single palmar crease
- Small lower jaw
- Undescended testicle
- Mental retardation
- Hole, split, or cleft in the iris
- Skeletal abnormalities
- Close-set eyes, or eyes that are fused together (possible symptom)
Around 80% of babies with this disorder are likely to suffer from a congenital heart defect. The forms of cardiac defects that children with this disease may have, include:
Ventricular septal defect
It involves an aperture (opening) between the lower chambers of their heart that prevent the heart from pumping blood in a normal manner.
Atrial septal defect
It is characterized by an opening between the two upper heart chambers that makes it difficult for the heart to pump enough oxygenated (oxygen-rich) blood to the various tissues of the body.
Patent ductus arteriosis
It is another type of cardiac defect in which a channel known as ductus arteriosus, which usually closes around the time of birth, fails to close.
Dextrocardia
In this form of defect, the heart of a child is located on the right side of the chest. Occasionally, this can give rise to acute heart defects.
Some typical birth defects associated with this disorder include the following:
- Holoprosencephaly
- Kidney defects
- Omphalocele
- Skin defects of the scalp
Infants and young children with this condition commonly also suffer from:
- Seizures
- Hypertension
- Kidney defects
- Gastroesophageal reflux
In some cases, they may also experience developmental disabilities and Scoliosis (an unnatural lateral curve to the vertebral column).
Patau Syndrome Causes
Babies are usually born with 46 chromosomes, arranged in 23 pairs. Children born with Patau Syndrome have an extra copy of chromosome 13 in every cell of their body. The presence of additional genetic material from chromosome 13 may occur due to:
Trisomy 13
Also known as Trisomy D, it is a condition which each cell of the body comprises of a full additional copy of chromosome 13. In a “Partial Trisomy”, only a part of this additional chromosome is present in the cells of the body.
As aforesaid, Patau Syndrome is often referred to as Trisomy 13.
Robertsonian translocation
In this condition, each cell consists of an additional partial copy of the chromosome.
Mosaicism
In this condition, an additional chromosome 13 can be found only in some of the cells of a sufferer.
The majority of people with this condition do not inherit it. Events leading to the development of the syndrome occur in either the sperm or the egg from which the fetus forms.
Most individuals with this disease have three copies of chromosome 13 in each of their body cells, instead of only two – as can normally be found. The additional genetic substance is disruptive to their course of development and gives rise to the characteristics of Patau Syndrome. The physical characteristics of patients with Mosaic Patau Syndrome are much milder in nature than sufferers of full Patau Syndrome.
Patau Syndrome Risk Factors
People with a personal or close family history of delivery of a child with this syndrome are at a greater risk of having it. Increase in maternal age also increases the susceptibility to this disorder, although the risk is not as pronounced as in Edward’s Syndrome or Down Syndrome.
Unless one of the parents happen to be a carrier of a translocation, the risk of having another child with this disease is less than 1%. This is even lower than that of Down Syndrome – a congenital disease arisng due to the presence of an additional 21st chromosome.
Patau Syndrome Diagnosis
The diagnosis of this disease is based on the signs and symptoms exhibited by sufferers. A single umbilical artery may be evident at birth in infants with this condition. In many cases, such patients exhibit signs of congenital heart disease, such as:
- Atrial septal defect
- Ventricular septal defect
- Patent ductus arteriosus
- Abnormal placement of the heart towards the ride side of the chest
Rotation of the internal organs of affected infants may be revealed with the aid of gastrointestinal x-rays or ultrasound examinations. Ultrasounds of the heart (Echocardiogram) are very important due to high frequency of cardiac defects related with this genetic disorder.
MRI or CT scans of the head can show problems in the brain structure. Such imaging exams can reveal problems like “Holoprosencephaly” which is characterized by fusion of both sides of the brain of a suffering infant. Apart from these exams, chromosome studies can help make a definitive diagnosis of Patau Syndrome as well as distinguish between full and partial forms of the condition and also Mosaicism.
Patau Syndrome Differential Diagnosis
Infants with Edward’s Syndrome and Patau Syndrome can exhibit similar features and make it difficult for physicians to differentiate. Naturally, the differential diagnosis should aim at properly distinguishing between the symptoms of these two conditions and confirming the presence of Patau Syndrome. A mild-pregnancy ultrasound scan can reveal certain abnormalities. However, further tests like chorionic villus sampling (CVS) or Amniocentesis are required to confirm the presence of the disorder.
Patau Syndrome Treatment
The treatment of children affected by this disorder involves planning on an individual basis. The forms of cure administered to a sufferer are based on his or her particular condition. The treatment for this disorder usually aims at addressing the specific physical symptoms that affected child are born with. Many infants find it difficult to survive in the first few days or weeks after birth due to complex heart defects or acute neurological problems.
In some cases, surgery may be needed to repair cleft palate, cleft lip or heart defects. Surgical intervention is typically held back for the first few months after birth due to the high mortality rate associated with the disease.
Occupational, physical and speech therapy can help Patau syndrome patients attain their complete developmental potential.
Patau Syndrome Complications
The possible complications associated with this condition include:
- Deafness
- Seizures
- Heart failure
- Vision problems
- Feeding problems
- Breathing difficulties or lack of breathing
The complications due to this disorder begin almost immediately after birth, with many newborns suffering from heart disease.
Patau Syndrome Prognosis
The disorder has been associated with many life-threatening complications. More than 80% of patients do not survive beyond the first month of their lives. Over 80% of sufferers do not live for more than the first year of their life. Only 5-10% of children with this condition manage to live past the first year of their life. In a study conducted on 21 individuals with this condition, only one managed to live up to 21 years of age while the others only survived past 5 years of age.
However, children affected with the partial or mosaic forms of this disorder may have a fully different outcome and hence – a much better prognosis.
Patau Syndrome Prevention
The prevention of this disorder is virtually impossible as it arises due to genetic factors. Unfortunately, most babies are lost at the time of pregnancy due to the acute complications associated with this disease.
However, couples with a personal or family history if the disorder can benefit from genetic counseling that can help them determine the risk of having a child with Patau Syndrome.
Screening tests conducted in the first trimester may indicate Patau or other abnormal chromosomal conditions. Through a process known as Amniocentesis, chromosome studies of amniotic cells can detect Trisomy 13 prior to birth.
Patau Syndrome – Support Group
Family members of patients of this disease can get in touch with the support group Support Organization for Trisomy 18, 13 and Related Disorders (SOFT) for necessary information and guidance about this disease. The contact details for this organization is as under:
SOFT President
Barb Vanherreweghe
2982 South Union Street
Rochester, NY 14624
Phone: (585) 594-4621
Toll-Free: (800) 716-SOFT (7638)
Children affected by this disorder and still managing to survive should be given the same care that other babies receive, which include visual and hearing assessments by 6-8 months of age as well as immunizations. Health problem should be treated according to their severity.
Patau Syndrome Pictures
Check out the following images that show the appearance of children affected by this syndrome.
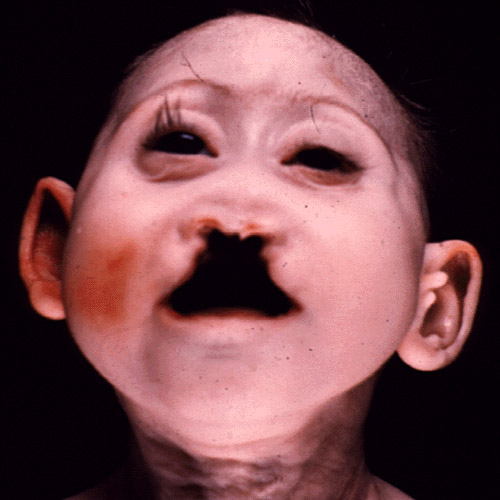
Picture 1 – Patau Syndrome
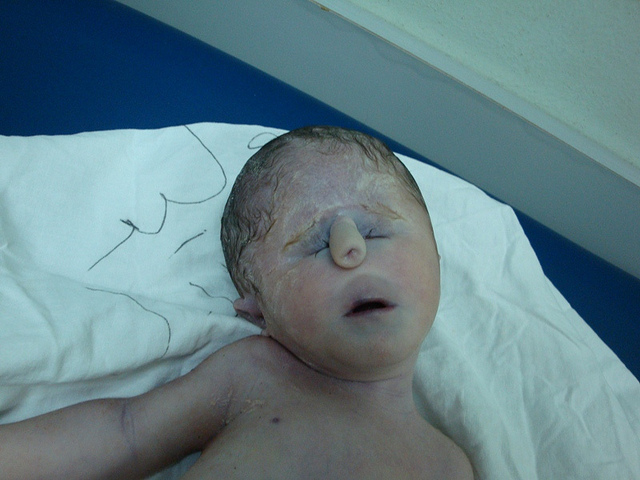
Picture 2 – Patau Syndrome Image
Those already with a child affected with this disorder and planning another baby should get in touch with a healthcare provider. Genetic counseling can help would-be parents understand the disease, the risk of inheriting it and also the ways to care for a suffering newborn.
References:
http://www.disabled-world.com/disability/types/patau-syndrome.php
http://rarediseases.about.com/od/rarediseasesp/a/patau05.htm
http://emedicine.medscape.com/article/947706-overview
http://emedicine.medscape.com/article/947706-overview#a0199
The post Patau Syndrome appeared first on Prime Health Channel.
]]>The post Tuberous sclerosis appeared first on Prime Health Channel.
]]>Tuberous sclerosis Definition
It refers to a group of two genetic conditions that leads to the growth of many benign (noncancerous) tumors in a number of regions in the body, such as:
- Brain
- Skin
- Heart
- Kidneys
- Nervous system
The disorders are named after a root or tuber-shaped growth in the brain.
The condition is also referred to by various other names, such as Tuberous sclerosis complex (TSC) and Adenoma sebaceum.
Tuberous sclerosis ICD9 Code
The ICD9 Code of this disease is 759.5.
Tuberous sclerosis Incidence
In the United States, the condition has a birth incidence in one out of every 6,000 individuals. It has a prevalence of one out of every 10,000 people.
The disorder affects all races and does not show a prominent racial predominance. It affects both sexes equally. According to some studies, males are more likely to suffer from neurological morbidity. However, this has not been conclusively demonstrated. TSC can arise at any age.
Tuberous sclerosis Symptoms
The signs and symptoms of this disorder involve:
Skin symptoms
These include:
- Red patches over the face, that comprise of many blood vessels (Adenoma sebaceum)
- Whiteness of various skin regions and having an appearance like confetti or an ash leaf
- Elevated skin patches with an orange-peel texture (shagreen spots), frequently arising on the back
Brain symptoms
These involve:
- Seizures
- Intellectual disability
- Delays in development
There can also be some other problems like:
- Pitted tooth enamel
- Benign, rubber-textured tumors arising on or around the tongue
- Development of rough growths around or under the nails of the fingers and toes
Tuberous sclerosis Causes
The cause of this condition is heredity. This is an inherited disorder. Mutations or changes in two genes, TSC1 and TSC2, lead to the development of this disease. These genes are usually supposed to prevent very fast or uncontrolled growth of cells. Mutations occurring in either the TSC1 or the TSC2 gene can cause excessive division of these cells, resulting in the growth of many lesions throughout the body of sufferers. Researchers are yet in the dark regarding the exact causes of these genetic mutations.
A child can get affected by this disease by getting the mutated gene from only one parent. The majority of cases of this disorder arise as a result of new mutations. Due to this reason, there is generally no family history of the condition.
The condition belongs to a group of disorders known as Neurocutaneous syndromes. Both the central nervous system (CNS) and the skin are involved.
Tuberous sclerosis Risk Factors
There are no known risk factors associated with this condition, other than having a parent with it. In such cases, each child has a 50% risk of inheriting the disorder. A person suffering from the disease has a 50% chance of passing it on to his or her biological children. The severity of the disorder may vary in nature. An individual having a mild form of the disease may have a child with a more acute form of the condition.
Approximately one-third of all individuals with the disease inherit a mutated TSC1 or TSC2 gene from a parent having the disorder. These are genes related to TSC. Around two-thirds of individuals with TSC have a new mutation in the TSC1 or the TSC2 gene.
Tuberous sclerosis Diagnosis
A child affected by this disorder is likely to be evaluated by different specialists who are trained to detect and cure problems of the areas where the tumors develop. The following doctors and specialists may be approached and consulted in case of tumors arising on the following regions of the body:
- Eyes (Ophthalmologist)
- Heart (Cardiologist)
- Brain (Neurologist)
- Skin (Dermatologist)
- Kidneys (Nephrologist)
In case a child reports of seizures, diagnostic testing is likely to include:
Electroencephalogram (EEG)
This exam records electrical activity in the brain and can help accurately determine the exact cause of the seizures in children.
In order to detect abnormal lumps on the kidneys and brain, diagnostic testing is likely to involve the following exams:
Ultrasound
This exam involves use of high-frequency audio-waves to create images of body parts like kidneys on a monitor.
Computerized Tomography (CT) Scan
It is an X-ray technique that creates images of the brain or other areas of the human body. The images produced in this scan are more detailed than those that are produced by standard X-ray tests.
Magnetic Resonance Imaging (MRI)
This exam makes use of radio waves and a magnetic field to produce cross-sectional images of the brain or other regions of the body.
In order to assess whether or not the heart of a child is affected, diagnostic testing is likely to include:
Electrocardiogram (ECG)
This exam records the electrical activity of the heart.
Echocardiogram
The test makes use of audio waves to produce images of the heart.
Doctors are also likely to carry out a physical examination. The skin and eyes of children would be examined thoroughly for lesions that are commonly related with TSC.
If a child is detected with TSC without a family history of the disease, parents may also consider screening for the condition. Monitoring and follow-up care is essential, even for mild cases of TSC that were undetected earlier.
Tuberous sclerosis Treatment
There is no particular treatment for this condition. As the disorder can differ from one person to another, treatment is based on the signs and symptoms of patients. Medical cure aims at the management of the symptoms.
The treatment options for the disorder include:
Psychological therapy
Consultation with a mental health therapist may help patients or their children adjust and accept to living with the condition.
Occupational therapy
People suffering from TSC can improve their ability to handle daily tasks through occupational therapy.
Educational therapy
Early therapeutic intervention can help affected kids overcome delays in development and meet their full potential in school.
Medications
Anti-epileptic medicines may be prescribed by doctors to control seizures. Other drugs may be administered by doctors to help manage problems in behavior. A medicine known as Everolimus (Zortress, Afinitor) is used to cure some forms of brain growths that cannot be removed in TSC patients with operation.
Another drug that may prove to be useful for brain growths and other problems resulting from TSC is an immunosuppressive drug known as Sirolimus (Rapamune). A topical application of this medicine may help cure the skin lesions (resembling acne) associated to TSC. However, Sirolimus is still being assessed in clinical trials and considered to be an experimental curative option as a treatment for TSC.
Surgery
Surgical removal may be opted for if a lesion affects the ability of a particular organ, like the kidney. In some patients, operation can help control seizures resulting from brain lesions that fail to respond to drugs. Surgical methods like laser treatment or Dermabrasion may improve the appearance of the lesions arising over the skin of sufferers.
Tuberous sclerosis Prognosis
Children affected by mild forms of the disorder generally do well. However, children with uncontrollable seizures or acute mental disability generally have a poor outcome. In some cases, when a child is born with severe TSC, one parent is found to suffer from a mild case of the disease that was undiagnosed.
In TSC patients, the tumors tend to be benign. However, some of the tumors (such as those on the brain or the kidney) may turn malignant.
Tuberous sclerosis Complications
The complications of the disorder may vary in nature, based on the location of development of the lesions. The lesions in the lungs can cause failure of the organ. Those in the kidney can result in its failure. However, it is only rarely that kidney lesions can turn cancerous in form.
Eye lesions can interfere with vision if they block a large section of the light-sensitive tissue situated at the retina (or the back of the eye). However, this only occurs in rare cases.
Cardiac lesions can obstruct blood flow or lead to Dysrhythmia (problems with heart rhythm). These growths are generally the largest at the time of birth and shrink with advancing age of children.
Brain lesions referred to as Subependymal giant cell astrocytomas or SEGA can obstruct the flow of cerebral spinal fluid inside the brain. This obstruction can manifest itself in the form of various symptoms, which include behavioral changes, headaches and nausea.
TSC can also lead to other problems like uncontrollable seizures and severe intellectual disability.
Tuberous sclerosis Prevention
Couples having a family history of the disorder and planning pregnancy can derive benefit from genetic counseling. Prenatal diagnosis is available for those families that are known to have a history of the disease. TSC often develops as a result of a new DNA mutation and these cases cannot be prevented.
Tuberous sclerosis Support Groups
Life can be pretty challenging for TCS patients and their parents or family members. The condition is very difficult to predict and has a negative impact on the academic, physical and social abilities of a child. It can lead to mild problems in some sufferers and more acute difficulties in others. Common problems like emotional and social withdrawal, aggression, raging outbursts and repetitive behavior can be very difficult to cope up with. However, discovering and curing problems on an early basis would help maximize the chances of a good outcome for sufferers. Patients of TSC and their family members would find it very useful to get valuable information and assistance from any of the various support groups associated to the disorder.
Sufferers can contact the Tuberous sclerosis Alliance at 800-225-6872. The Tuberous sclerosis Association can be reached at 01332 250734. Parents may also get in touch with the doctor of their child and know about any local forums or support groups that offer assistance.
Tuberous sclerosis Pictures
The following images help you understand the physical appearance of TSC patients.
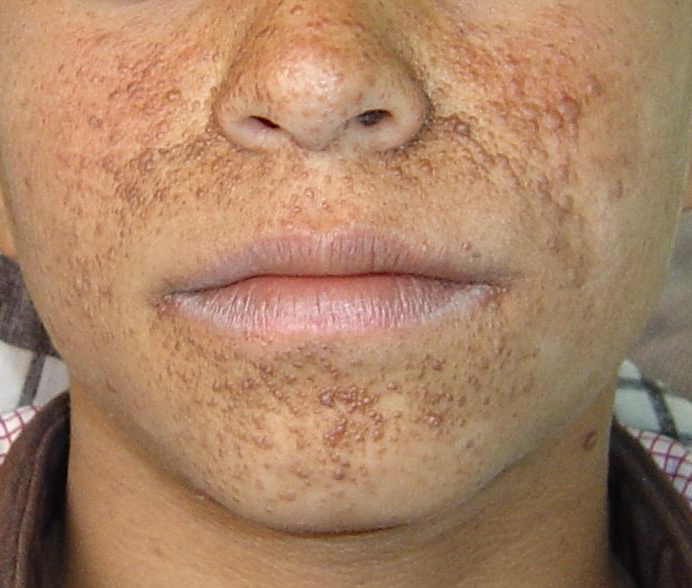
Picture 1 – Tuberous sclerosis
Picture 2 – Tuberous sclerosis Image
Although Tuberous sclerosis does not have any cure, its symptoms can be managed with the aid of proper medical treatment. If you or any of your family members is suffering from the symptoms of TSC, it is important that you seek diagnosis and medical treatment on an immediate basis.
References:
http://www.mayoclinic.com/health/tuberous-sclerosis/DS01032
http://www.nlm.nih.gov/medlineplus/ency/article/000787.htm
https://en.wikipedia.org/wiki/Tuberous_sclerosis
http://emedicine.medscape.com/article/1177711-overview
The post Tuberous sclerosis appeared first on Prime Health Channel.
]]>The post Thalassemia appeared first on Prime Health Channel.
]]>What is Thalassemia?
It is an inherited blood disease in which the body produces an abnormal type of hemoglobin – the protein present in the red blood cells that transports oxygen. In patients of Thalassemia, there is excessive destruction of RBCs (red blood cells) which results in anemia.
The condition is referred to by various other names, such as:
- Cooley’s anemia
- Alpha Thalassemia
- Beta Thalassemia
- Mediterranean anemia
Thalassemia ICD9 Code
The ICD9 Code for this disorder is 282.4.
Thalassemia Types
The condition is mainly differentiated into the following two types:
Alpha Thalassemia
It arises when one or more genes associated to the alpha globin protein are either changed or missing. It most commonly occurs in individuals from China, Middle East, southeast Asia and Africa.
Beta Thalassemia
It develops when similar genetic impairments affect the manufacture of the beta globin protein. It occurs in people of Mediterranean descent and to a lesser extent, Asians, Chinese and African Americans.
Both Alpha and Beta forms of Thalassemia can be classified into the following two subtypes:
Thalassemia Major
This type develops when an individual inherits the impaired gene from both parents. Beta Thalassemia Major is also known as Cooley’s anemia.
Thalassemia Minor
It arises if a person gets the impaired gene from only one parent. People with this type of the disease are carriers of the condition and generally do not experience any signs and symptoms.
Thalassemia Symptoms
The signs and symptoms of this disorder may include the following problems:
- Irritability
- Dark urine
- Fatigue
- Weakness
- Slow growth
- Abdominal swelling
- Pale appearance
- Shortness of breath
- Facial bone deformities
- Yellow discoloration of skin (indicative of Jaundice)
The symptoms experienced by sufferers are based on the form and severity of the condition. Some babies may manifest the symptoms at birth while others may develop its signs during the first two years after birth. Certain individuals suffering from only one affected hemoglobin gene do not experience any symptoms of Thalassemia.
Alpha Thalassemia Symptoms
Four genes are associated in the production of the alpha hemoglobin chain. Patients receive two genes from each parent. The signs and symptoms may differ on the basis of the number of genes acquired by patients:
- Patients do not manifest any signs of Thalassemia in case of only one mutated gene. However, a carrier of the disorder can pass the gene on to children.
- The signs are mild in case of two mutated genes. This form of the disorder may be referred to as Alpha- Thalassemia minor. Patients may also be said to show an Alpha- Thalassemia trait.
- The symptoms may range from moderate to severe in case of three mutated genes. This type of the disorder is also known as Hemoglobin H disease.
- In case of four mutated genes, the disease is referred to as Hydrops fetalis or Alpha-Thalassemia Major. It generally leads to the death of a fetus prior to delivery or right after birth.
Beta Thalassemia Symptoms
Two genes are associated in the manufacture of the beta hemoglobin chain. A person may acquire one gene from each parent. The symptoms may depend on the number of genes received by sufferers:
In case of one mutated gene, sufferers may have mild symptoms. This type of the disease is known as Beta- Thalassemia minor or called a Beta-Thalassemia trait.
Acquiring two mutated genes may lead patients to suffer from symptoms that range from moderate to severe in nature. The form of the disorder is referred to as Cooley’s anemia or Beta-Thalassemia major. Babies who are born with two impaired beta hemoglobin genes are generally healthy at birth. However, they develop the signs within the first two years after birth.
Thalassemia Causes
The disorder arises due to mutations in the DNA of those cells that manufacture hemoglobin, the protein transporting oxygen in the body. The mutations that are related with Thalassemia are passed on to children from parents.
The mutations resulting in Thalassemia disrupt the normal production of hemoglobin and lead to low levels of the protein. As a consequence, this results in a high rate of destruction of red blood cells and leads to anemia. In anemic individuals, there are not enough amounts of red blood cells (RBCs) to transport oxygen to the tissues. This leaves sufferers fatigiued.
Thalassemia Risk Factors
The factors that increase the risk of suffering from this condition include:
Being of certain origin
The condition most often arises in individuals of Greek, Asian, African, Middle Eastern and Italian ancestry.
Having a family history
The disease is passed on to children from parents through mutated hemoglobin genes. Having a family history of the condition can elevate the risk of the disorder in individuals.
Thalassemia Diagnosis
The majority of children exhibiting moderate to severe form of Thalassemia exhibit the symptoms within the initial two years after birth. The diagnostic tests for this disorder may include:
Physical examination
A physical examination may reveal an enlargement of the spleen.
Blood tests
If a physician suspects the presence of Thalassemia, he or she may recommend blood tests to confirm the diagnosis.
In case of the presence of Thalassemia, blood tests may show:
- Paleness of RBCs (Red blood cells)
- Low level of RBCs
- Smaller appearance and abnormal shape of red blood cells under microscopic examination
- Uneven distribution of hemoglobin in red blood cells, which gives a bull’s eye appearance to the cells under the microscope
Blood tests may also be used for the following purposes:
- Evaluation of the hemoglobin
- Measurement of the amount of iron in blood
- Performing DNA analysis to detect Thalassemia or to assess whether or not an individual is carrying mutated hemoglobin genes
Prenatal testing
Before the birth of a baby, this type of testing can be done to determine the presence of Thalassemia and how severe it is. Tests used to detect the condition in fetuses include:
Amniocentesis
The test is generally conducted around the 11th week of pregnancy and involves the removal of a small piece of placenta for assessment.
Chorionic villus sampling
The test is generally carried out around the 16th week of pregnancy. It involves extracting a sample of the fluid surrounding the fetus.
Assisted reproductive technology
This procedure combines pre-implantation genetic diagnosis along with in-vitro fertilization and may assist parents having Thalassemia or the carriers of an impaired hemoglobin gene who give birth to healthy infants. The method includes retrieving mature eggs from a woman and fertilizing them in a laboratory dish with a male sperm. The embryos are tested to determine the presence of impaired genes. Only those not having genetic impairments are implanted in the woman.
Thalassemia Differential Diagnoses
The differential diagnoses of this disorder include distinguishing its signs from those of disorders that give rise to similar symptoms:
- Leukemia
- Iron deficiency anemia
- Hemoglobinopathy
- Other types of Thalassemia
Doctors must ensure that the signs of the above disorders are not what patients suspected of Thalassemia are actually complaining about.
Thalassemia Treatment
The treatment for the condition depends on the type of the disorder that a patient suffers from as well as its intensity. In mild Thalassemia, symptoms are usually mild and little or no treatment is required. A blood transfusion may be needed in the following cases:
- After an operation
- After giving birth to a baby
- To help control the symptoms of the disease
The treatment for moderate to severe cases of the disease may involve:
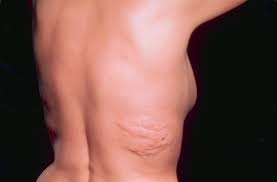
Stem cell transplant
The process is also referred to as “Bone Marrow Transplant.” In selected cases, it may be used to cure severe Thalassemia. Before this procedure, extremely high doses of radiation or drugs may be needed to destroy the diseased bone marrow in sufferers. Following this, patients may receive stem cell infusions from a compatible donor. However, this method can involve acute risks for sufferers which can also include death. Due to this reason, Stem cell transplant is generally reserved for patients with the most acute form of the disorder having a compatible donor – generally a sibling.
Frequent blood transfusions
Frequent blood transfusions are often needed for more acute forms of Thalassemia, possibly in every few weeks. However, this may cause buildup of excess iron in the body and medicines may be needed to get rid of it.
Thalassemia Prognosis
Acute cases of this disorder can lead to early death of sufferers as a result of cardiac failure, generally between 20 and 30 years of age. Regular blood transfusions and therapy can help remove excess iron from the body and improve the outcome of the disease.
The lifespan is generally normal in less severe types of the disease.
In people with a family history of the disorder who are planning to have children, prenatal screening and genetic counseling can be helpful.
Thalassemia Complications
If left untreated, the condition can result in cardiac failure and liver problems. It can also make patients susceptible to infections. Some of the symptoms can be managed with blood transfusions although they may lead to high levels of iron in the body which can cause damage to the endocrine system, liver and heart.
Thalassemia Prevention
The disorder cannot be prevented in the majority of cases. In people having Thalassemia or carrying a gene responsible for the disease, consultation with a genetic counselor can provide patients with valuable guidance before having a child.
Thalassemia Coping and Support
Coping with this disorder can be quite challenging. However, patients can find useful information, guidance and assistance with the various support groups and forums existing online and off the web. You can consult your doctor to know about support groups in your area or get in touch with the Cooley’s Anemia Foundation at 800-522-7222.
Thalassemia Pictures
The images below can help you get a visual idea about the disorder.
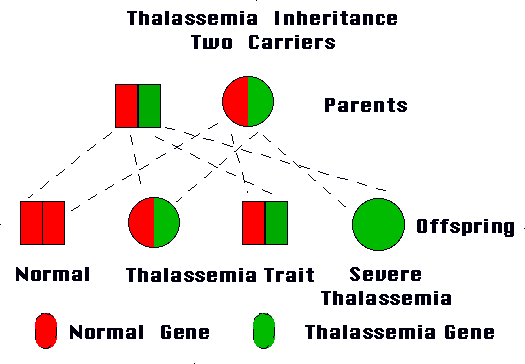
Picture 1 – Thalassemia
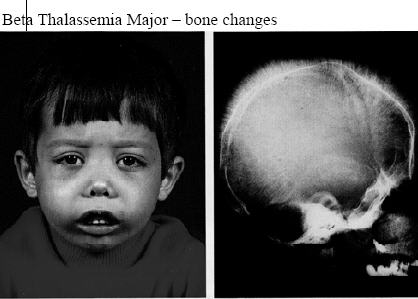
Picture 2 – Thalassemia Image
If you suspect yourself or your child to be having the symptoms of Thalassemia, get in touch with a professional medical care provider as soon as possible. Left untreated, the disorder can wreak havoc on your system and leave you susceptible to various problems some of which can even be life-threatening. Naturally, it is best to seek early diagnosis and treatment which can ensure better management of the problems.
References:
http://www.mayoclinic.com/health/thalassemia/DS00905
http://www.nlm.nih.gov/medlineplus/ency/article/000587.htm
http://www.umm.edu/ency/article/000587.htm
http://www.mdguidelines.com/thalassemia
http://en.wikipedia.org/wiki/Thalassemia
The post Thalassemia appeared first on Prime Health Channel.
]]>